烯烃硼氢化氧化(具有动力学立体控制的取代环己烷的模块化合成)
Posted
篇首语:早晨要撒你种,晚上也不要歇你手。本文由小常识网(cha138.com)小编为大家整理,主要介绍了烯烃硼氢化氧化(具有动力学立体控制的取代环己烷的模块化合成)相关的知识,希望对你有一定的参考价值。
烯烃硼氢化氧化(具有动力学立体控制的取代环己烷的模块化合成)
具有动力学立体控制的取代环己烷的模块化合成
文章出处:Yangyang Li, Yuqiang Li, Hongjin Shi, Hong Wei, Haoyang Li, Ignacio Funes-Ardoiz, Guoyin Yin. Modular access to substituted cyclohexanes with kinetic stereocontrol. Science 2022, 376, 749-753.
摘要:取代六元环烃是生物活性化合物的常见组成部分。虽然二取代环己烷的合成方法在热力学上是有利的,但合成它们的立体异构体的可靠和模块化的协议仍然是难以捉摸的。在此,作者报道了一种通过实施链行走催化,由易于接近的取代甲基环己烷模块化合成具有良好动力学立体控制的二取代环己烷的一般策略。力学上,在环己烷附近引入一个立体要求高的硼酯基是指导立体化学结果的关键。与目前的交叉偶联技术相比,这种方法的合成潜力在复杂生物活性分子的后期修饰中得到了强调。
与平坦的生物同位体相比,药物分子的三维复杂几何结构通常赋予其更好的生物活性和物理特征。因此,建立高效的合成方案以立体定向方式构建饱和环引起了人们的极大兴趣。一个取代基位于赤道位置,另一个取代基位于轴向位置,即1,2-顺式,1,3-反式和1,4-顺式取代模式(图1A),代表了广泛的药物分子中的关键骨架,如抗癌3期药物林罗多司他;组织蛋白酶S抑制剂;羟固醇受体LXR beta;组胺H3R ADS10227 (图1B)。此外,它们往往表现出比它们在热力学上有利的同分异构体更好的生物活性。尽管自Diels-Alder反应的发现以来,关于环己烷骨架结构高效合成方法的研究并未停止,但合成热力学上不受欢迎的取代环己烷的模块化和可变方法仍然缺乏。传统的环己基衍生物交联为制备取代环己烷提供了可靠的平台,但这些方法均有利于热力学稳定的立体异构产物,即1,2-反式、1,3-顺式和1,4-反式异构体。三取代烯烃的选择性加氢或1,1-二取代烯烃的氢化是获得这些化合物的另一种途径。然而,在这些过程中控制面选择性仍然是一项具有挑战性的任务(图1C)。虽然过渡金属催化的取代芳烃加氢是合成全顺式取代环己烷的一种特别有吸引力的方法,但这种方法不能合成具有1,3-反式取代基的环己烷。鉴于环己烷骨架的普遍使用和目前合成方法的局限性,开发热力学上不受欢迎的二取代环己烷的高立体选择性和高效的模块化合成策略,不仅可以极大地丰富有机合成化学家的工具箱,而且可以扩大化合物库,用于药物发现。
受金属氢化物试剂尺寸对取代环己酮还原的立体选择性的启发,以及作者之前对烯烃迁移二官能化的研究,作者推测,如果一个体积较大的基团可以加入到一个取代的亚甲基环己烷的外环位置,它将生成一个具有空间要求的三取代烯烃,在催化剂的作用下能够增强立体分辨能力(图1C)。随后与亲电试剂偶联得到了热力学上不利的具有良好对映体比例的二取代环己烷。在这里,作者报道了其成功地实现了这一概念,通过在Ni催化的迁移碳硼化反应中使用取代的亚甲基环己烷作为偶联配体,将可转化的硼基原位结合(图1D)。硼官能团的安装不仅在控制立体选择性方面起着重要作用,而且通过众所周知的C-B键功能化化学,为进一步的后期功能化生成一个关键的反应位点。
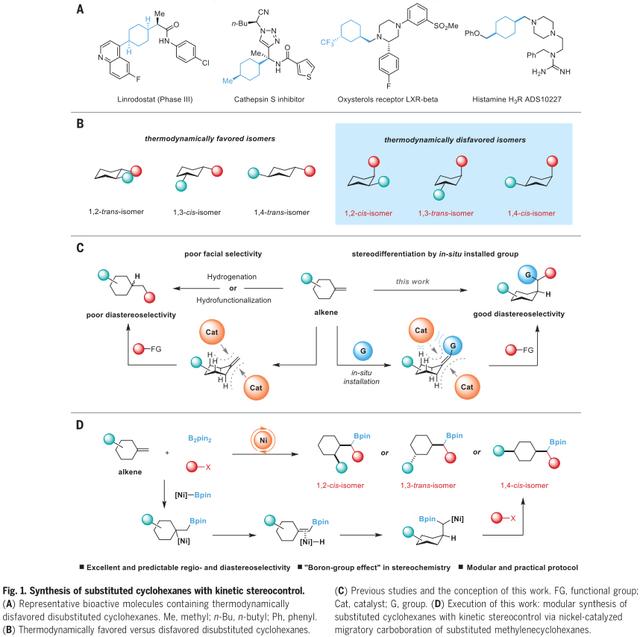
图1
作者通过选择末端烯烃化合物1,双(pinacolato)二硼(B2pin2) (化合物2)和有机卤化物3作为模型底物开始了这项研究。配体评价表明,该反应的化学选择性高度依赖于配体骨架,位阻吡啶基恶唑啉(PyrOx)配体能有效促进1,4-顺式产物4的形成(图2A)。在仔细检查了每个反应参数后,作者确定了乙酰丙酮镍[Ni(acac)2]和PyrOx配体化合物L6作为预催化剂,甲醇锂(LiOMe)为碱,N-甲基-2-吡咯烷酮(NMP)/1,4-二氧六环(9: 1)为溶剂,35 oC为最佳反应条件。使1,4-顺式产物4具有优良的顺/反选择性(> 99: 1)。相反,当B2pin2被频哪醇甲硼烷(HBpin)取代时,反马氏烷基化产物8的产率为30%,但其对映选择性较差(顺式/反式 = 5: 1) (图2B),而烯基硼化合物5的氢烷基化反应保留了高水平的对映选择性(顺式/反式 = 83: 1) (图2C)。反应效率的降低是由烯烃异构化引起的,这可能是由于在催化剂活化过程中形成了意想不到的Ni(I)-H物种所致。这些结果清楚地支持了频哪醇硼酸酯(Bpin)基团的初始加入对提高反应的立体选择性起着重要作用的假设。为了进一步证实这一效应,作者对Ni-H迁移插入步骤进行了密度泛函理论计算(图2D)。仔细分析了Ni-H插入烯烃化合物1的不同赤道构象和轴向构象的过渡态后的结果表明,由于PyrOx配体与硼官能团之间存在较大的空间位阻,顺式迁移插入的能量为3.7 kcal·mol-1。尽管氢的体积很小,但过渡态的局部空间位阻由整个[Ni]-H体系产生,有利于氢原子在赤道面上的安装,Bpin基团和PyrOx配体放置在较远的距离(对1,3插入观测到相同的效果)。相反,在没有Bpin基团的情况下,由于配体在轴向和赤道位置具有较大的构象灵活性,两个过渡态的立体分化程度都较低,几乎是等能的。
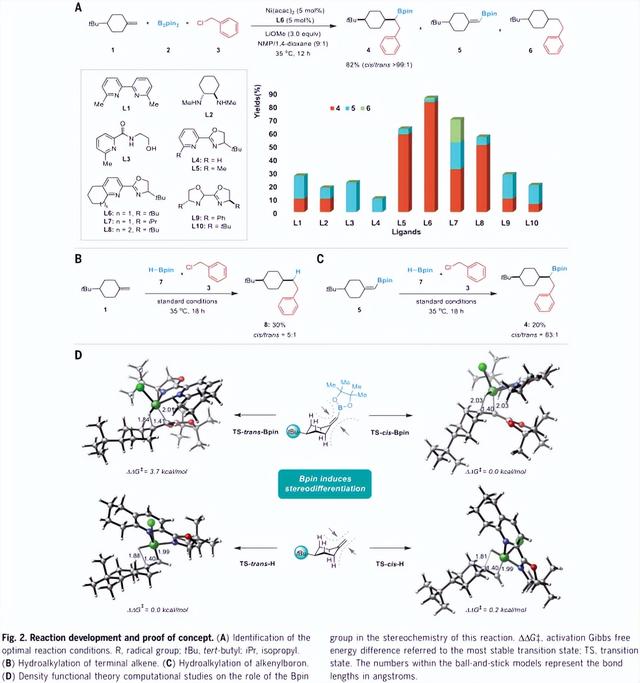
图2
接下来,作者关注Ni催化反应的普遍性,首先是一系列4-取代的亚甲基环己烷(图3A)。当多种取代基包括烷基、酯和芳基(化合物9-25和29-31)以及杂原子(化合物26-28)被连接时,相应的仲烷基硼酯具有良好的1,1-区域选择性和1,4-顺式立体选择性(91: 9-99: 1 dr),产率从适中到良好。值得注意的是,即便使用较小的甲基取代基(化合物32),也可以获得良好的对映选择性(dr = 91: 9)。此外,所得到的环己烷骨架(化合物20和23-25)在液晶材料中有广泛的应用。烷基卤化物芳烃环上取代基的变化不影响反应效率和对映选择性。接下来,作者检测了含有3取代基的亚甲基环己烷,得到了良好的1,3反式-对映选择性(图3B)。此外,从2-取代的亚甲基环己烷中得到了同样高的对映选择性的1,2-顺式环己烷衍生物(图3C)。除六元碳环外,杂环哌啶衍生物(化合物42-44)也与所开发的方案相兼容,得到的动力学产物具有较高的面选择性(图3D)。双组分偶联产物或烯烃异构化产物构成了这些反应剩余的大部分质量平衡。产物的立体化学结构(化合物13、16、22、25、27、28、32和41)通过单晶X射线晶体学明确证实。在温和的反应条件下,芳基溴(化合物13)、酯(化合物16和19)、苯酚(化合物21)、氰基(化合物22)、缩醛(化合物25)、苄基醚(化合物26)、亚胺(化合物27)和酰胺(化合物28)等多种柔性官能团都能很好地耐受。
受上述结果的鼓舞,作者随后研究了具有多重取代亚甲基环己烷部分的结构复杂分子。如图3E所示,具有不同取代模式的八氢吲哚(化合物45)、石胆酸(化合物46)、诺卡酮(化合物47)、(-)-薄荷酮(化合物48)和屈他雄酮丙酸酯(化合物49)的配合物分子均通过该方案转化为单一产物异构体。此外,端烯烃与苯环相连的(−)-氧化石蒜(化合物50)、端烯烃为五元环的雌二醇二酮-3-酮(化合物51)、 端烯烃为无环的(+)-诺卡酮(化合物52)都能顺利地参与到Ni催化反应中,生成具有优异面选择性的相应产物(fs > 95:5)。这些结果表明了该方案在复杂生物活性分子后期多样化中的兼容性和潜力。
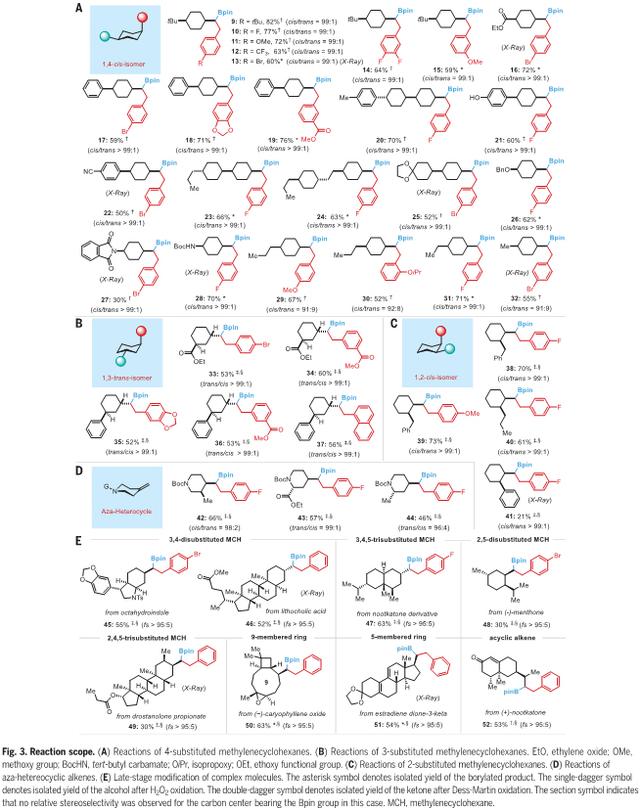
图3
硼官能团与分子的结合尤其具有吸引力,因为它们具有不同的下游转化。为了进一步说明该方法的合成潜力,对产品进行了额外的多样化。如图4所示,以cis-13为模型底物,碳硼键可以通过Matteson反应转化为C-C(sp3) (化合物53)或C-C(sp2) (化合物54)键,也可以通过Liu课题组的胺化方案转化为C-N键(化合物55)。此外,Pd催化的芳基溴部分的交叉偶联提供了与硼酯完整的胺化产物(化合物56)。因此,得到了几种结构多样的顺-1,4-二取代环己烷。此外,还可从产物中制备双环醚化合物57和杂双环化合物58。
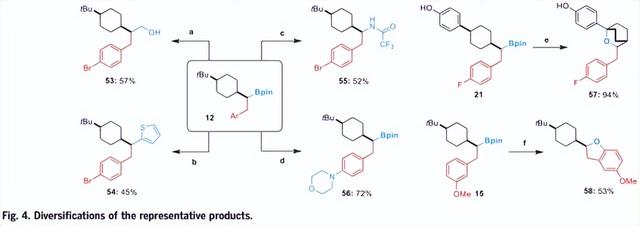
图4
为了突出该协议的价值,作者使用经典交叉偶联方法和作者的方法进行了比较研究。一般来说,环己酮(或环己醇)是很容易得到的化合物,可以通过还原、卤化和经典的交联反应得到热力学上有利的产物。相反,同样的酮可以经过烯烃化,然后通过该方案生成具有反向立体中心的类似物。特别值得注意的是,观测到的立体化学不仅存在于简单的双取代环产物(化合物cis-8)中,也存在于多取代环己烷分子(化合物60c和62c)中。这是对目前氢化技术(形成热力学上有利的产品)的补充。
通过这项工作,作者实现了Ni催化的化学选择性三组分偶联反应,该反应为从易于合成的亚甲基环己烷、B2pin2和苄基卤化物到热力学上不受欢迎的取代环己烷的框架提供了方便。广泛的官能团,包括芳基溴、酯、胺、酰胺、亚胺和α,β-不饱和酮,都与这种转化兼容。
相关参考